

Guest article: Exempt today, regulated tomorrow? The current state of play in the medical device industry
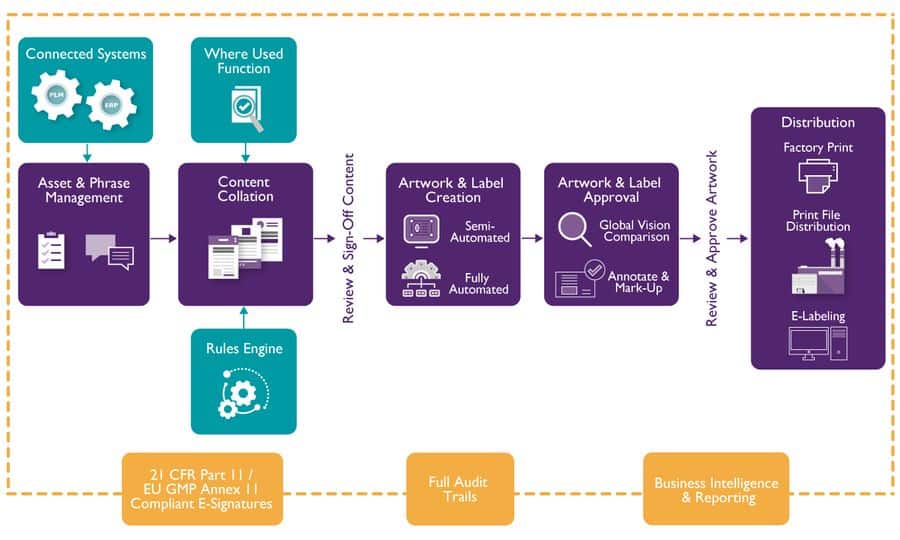
No sooner than EU Medical Device Regulation (MDR) had entered into force in May 2021 did the focus shift to the compliance deadline for its ‘sister’ regulation, IVDR. With this constant regulatory pipeline more likely to accelerate than subside, manufacturers need to ensure they have a full picture of rules – and how can they get ahead of the constant push for compliance. Bob Tilling, VP Global Sales at Kallik, understands the compliance pressures currently facing medical device firms, and is sure that compliance rules will only further tighten in the years ahead. Here he assesses the technologies that could help ease the pain of costly compliance projects.
In recent months, the number of companies seeking MDR compliance projects and solutions has noticeably dropped – a sign that these firms may be tentatively getting to grips with delivering MDR compliance throughout their business operations. But they must not rest on their laurels – there is bound to be more to come.
The sheer scale of the compliance effort for medical device manufacturers doing business in the European market was a major eye-opener for many business leaders. Asset siloes were uncovered, disparate systems identified, and labels and artwork painstakingly updated – often manually, at extensive cost.

Plenty of companies operating in the medical device market are doing a great job of using technology to handle assets such as labels and artwork that require management at both the global and national levels in response to regional rules and market requirements.

However, this often only extends to content management instead of full editing, version control and re-issuing that is often require in bulk to streamline compliance projects. It is clear there is more to be done for many firms in terms of addressing full digital maturity.
MDR should be seen as a wake-up call – not a one-off challenge
One Kallik client, a leading multinational medical device manufacturer, internally updated over 90,000 labels in just six months to achieve full MDR compliance – a monumental task that simply would not have been possible to achieve at scale without leveraging technology.
Yet many firms are still a long way from complying with the full extent of MDR rules such as updated product Instructions for Use (IFUs) – and the regulatory pace of change is not set to stop there. Here’s how the medical device regulatory landscape could evolve over the coming months and years – and how firms can prepare early to avoid costly, disruptive compliance burdens:
Exempt today, regulated tomorrow: The compliance scope is widening
Regulators in the EU, individual member states and further afield are all continually refining the requirements and scope of their regulations for the medical device market – both reactively in response to product deficiencies, or proactively to tighten quality and traceability.
Wider and widening still…
MDR, which replaced the previous EU Medical Devices Directive, is a case in point. The scope for devices that fall under MDR for requirements such as Unique Device Identifier labels has risen significantly, expanding from roughly 2.6 million devices to five million. Coloured contact lenses, dermal fillers, medical software – even AI solutions – now fall under the regulations, leaving manufacturers scrambling to comply with unfamiliar rules.
These firms dealing in products previously exempt or outside the scope of older legislation now run the risk of an uncomfortable, short-notice demand for compliance. Once regulators begin to flex their muscles and crack down on non-compliance where they identify risks, there could be significant pain on both a financial and reputational level.
IVDR is next in the pipeline
The challenges don’t stop there. IVDR, the counterpart legislation to MDR covering in vitro diagnostic medical devices, is due to come into force from May 2022.
Those firms that scraped compliance just in time for the 2021 MDR deadline now find themselves faced with yet another large compliance project, while specialist in vitro device firms may be unaware of the major burden and challenges posed by such regulations.
Beyond this, the UK’s MHRA announced a consultation in September 2021 with a view to establishing a new regulatory regime for the domestic market in the next few years. Once firms get to grips with MDR and IVDR, they could be immediately faced with a new UK-only independent regulatory framework, with a differing set of regulatory rules.
Medical device organisations face an uncertain yet fast-moving regulatory environment to contend with – and this could tighten even further.
What lies ahead: Regional rules, stricter serialisation?
Certainly in my two decades in the medical device space, the trend for compliance has only gone in one direction – more rules, covering more product lines, down to a more granular level. It is hard to see a departure from this trend in the coming months and years.
Going forwards, we can expect to see serialisation significantly ramped up – extending end-to-end traceability requirements from the current batch level even down to individual products such as a bandage or syringe within a larger package.
Looking at regulation at the national level, we can also expect compliance efforts to be duplicated due to the introduction of the UKCA product marking for the UK market, scheduled to replace existing CE markings from July 2023. If firms already have every device marked differently for sale in the U.S. and EU markets, this will have to be duplicated again if they wish to continue selling their products in the UK.
Technology will be critical to meet the growing compliance burden
Regardless of how the regulatory pipeline shapes up, one lesson to be learned from the struggles witnessed during the MDR compliance push remains clear – manual solutions will be a non-starter for successive rules and regulations.
Will organisations be able to manually identify, edit and re-issue labels, artwork and packaging affected by new regulations? Certainly not without investing excessive time, capacity and finances. Adopting an end-to-end digital solution such as Veraciti will be critical to provide effortless mass asset adjustments in response to emerging regional or national regulatory shifts.
If medical device organisations act now, they will reap the long-term rewards – all while benefitting from continuous operational efficiencies and powerful new artwork and label management capabilities.
https://thiis.co.uk/guest-article-exempt-today-regulated-tomorrow-the-current-state-of-play-in-the-medical-device-industry/https://thiis.co.uk/wp-content/uploads/2022/05/veraciti-diagram.jpghttps://thiis.co.uk/wp-content/uploads/2022/05/veraciti-diagram-150x150.jpgAnalysis & InsightsNewsroomOpinions & Commentscompliance,device,Kallik,manufacturers,MDR,medical,Mobility,regulationNo sooner than EU Medical Device Regulation (MDR) had entered into force in May 2021 did the focus shift to the compliance deadline for its ‘sister’ regulation, IVDR. With this constant regulatory pipeline more likely to accelerate than subside, manufacturers need to ensure they have a full picture of...Liane McIvorLiane McIvorliane@thiis.co.ukEditorTHIIS Magazine